Hardy-Weinberg Principle
The Hardy-Weinberg principle is a mathematical model used to describe the equilibrium of two alleles in a population in the absence of evolutionary forces. This model was derived independently by G.H. Hardy and Wilhelm Weinberg. It states that the allele and genotype frequencies across a population will remain constant across generations in the absence of evolutionary forces. This equilibrium makes several assumptions in order to be true:
- An infinitely large population size
- The organism involved is diploid
- The organism only reproduces sexually
- There are no overlapping generations
- Mating is random
- Allele frequencies equal in both genders
- Absence of migration, mutation or selection
As we can see, many items in the list above can not be controlled for but it allows for us to make a comparison in situations where expected evolutionary forces come into play (selection etc.).
Hardy-Weinberg Equilibrium
The alleles in the equation are defined as the following:
- Genotype frequency is calculated by the following:
- Allele frequency is calculated by the following:
- In a two allele system with dominant/recessive, we designate the frequency of one as p and the other as q and standardize to:
- Therefore the total frequency of all alleles in this system equal 100% (or 1)
- Likewise, the total frequency of all genotypes is expressed by the following quadratic where it also equals 1:
- This equation is the Hardy-Weinberg theorem that states that there are no evolutionary forces at play that are altering the gene frequencies.






Calculating Hardy-Weinberg Equilibrium (activity)
This exercise refers to the PTC tasting exercise. One can test for selection for one allele within the population using this example. Though the class size is small, pooling results from multiple section can enhance the exercise. Remember to surmise the dominant/recessive traits from the class counts.

- What is the recessive phenotype and how can we represent the genotype?
- What is the dominant phenotype and how can we represent the genotypes?
- What is the frequency of recessive genotype? (q2)
- What is the frequency of the recessive allele? (q)
- What is the frequency of the dominant allele?(p=1-q)
- Use Hardy-Weinberg to calculate the frequency of heterozygotes in the class. (2pq)
- Use Hardy-Weinberg to calculate the frequency of homozygotes in the class. (p2)
- Using an aggregate of multiple section, compare the local allelic and genotypic frequencies with what the Hardy-Weinberg would predict.
- With this small number in mind, we can see that there are problems with the assumptions required for this principle. The instructor will perform the following simulation in class to illustrate the effects on multiple populations with the effects of selection and /or population limitations. A coefficient of fitness can be applied to illustrate a selective pressure against an allele.
- In the case of a selective pressure, a fitness coefficient (w) can be introduced. A research article http://www.jci.org/articles/view/64240 has shown that the Tas2R38 receptor aids in the immune response against Pseudomonas. Imagine a situation where there is an epidemic of antibiotic resistant Pseudomonas. This would
show that the dominant allele will have a selective advantage.- Modify the fitness coefficient in the Population Genetics Simulator and describe the effects this would have over many successive generations.
A case study of evolution: Population Genetics at Work
Additional Resources/Experiments
- Population Genetics Simulation of Alleles (Schaffer)
- Population Genetics Simulation of Alleles (Sheehy)
Tags: quantitative reasoning, integration of knowledge
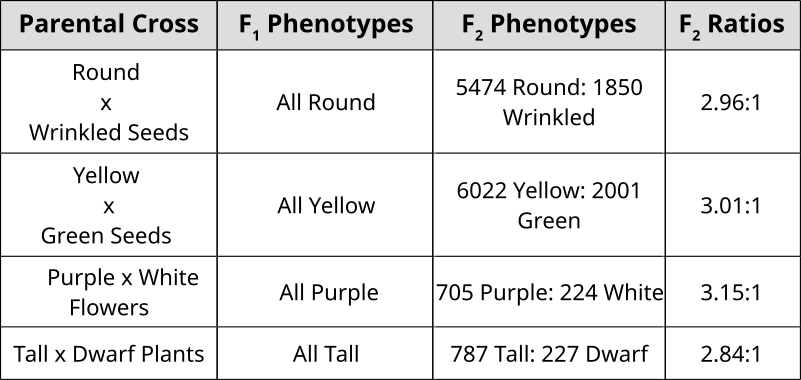

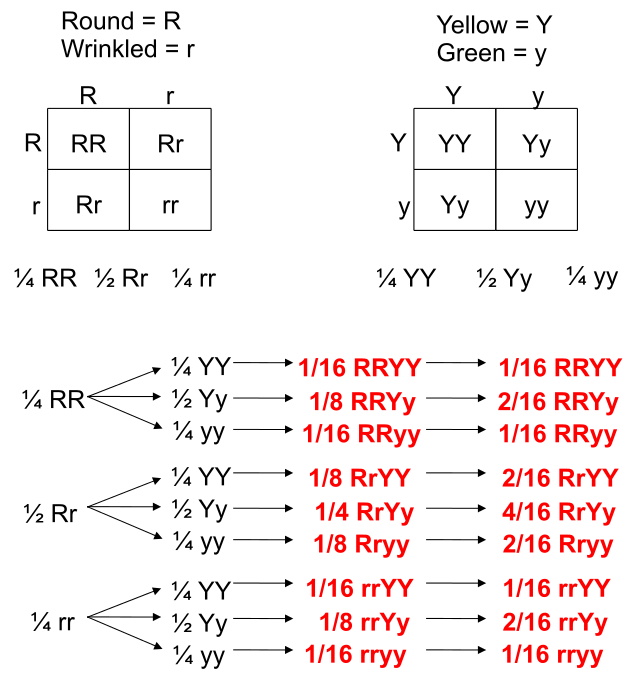









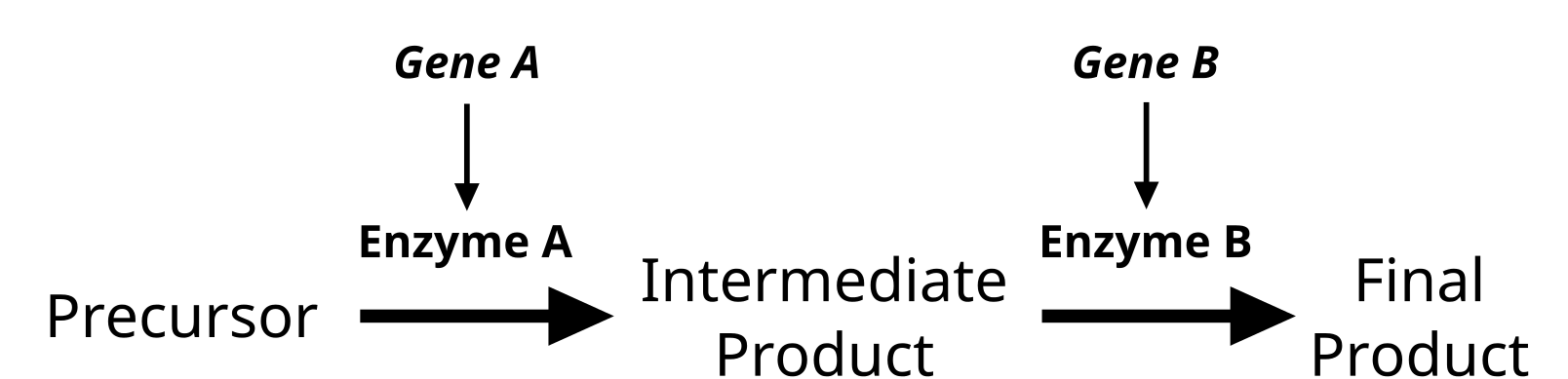



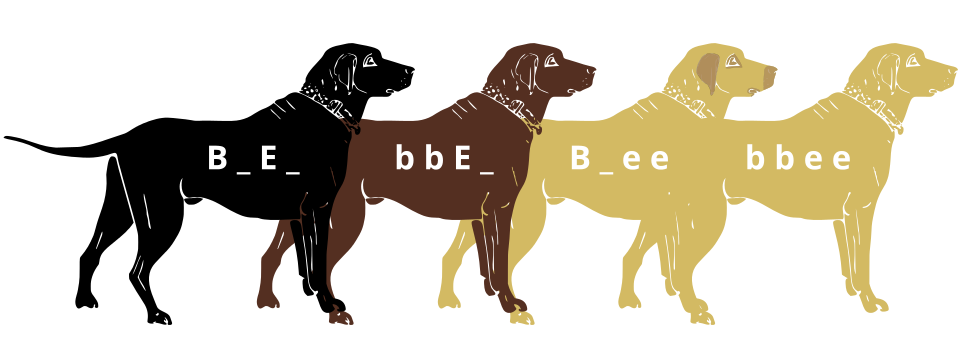











 [GFDL (http://www.gnu.org/copyleft/fdl.html) or CC-BY-SA-3.0 (http://creativecommons.org/licenses/by-sa/3.0/)], via Wikimedia Commons")











.svg)



