Enzyme kinetics of Turnip Peroxidase
Hydrogen peroxide (H2O2) is a strong oxidizing agent that can damage cells and is formed as a by-product of oxygen consumption. Fortunately, aerobic cells contain peroxidases that break down peroxide into water and oxygen. This enzyme reduces hydrogen peroxide into H2O by oxidizing an organic compound (AH2 -> A).

This exercise uses turnip extract as a source of peroxidase. This turnip extract requires a source of electrons (a reducing agent) in order for the reaction to occur. In this case, a colorless organic compound called guaiacol is used. Guaiacol is oxidized in the process of converting the peroxide and becomes brown. Enzymatic activity can then be traced using a spectrophotometer to measure the amount of brown being formed.
Hypotheses on Enzyme Kinetics
Use the following questions to develop your hypotheses based on the background understanding of enzymes. Discuss if the outcomes matched your hypotheses following these exercises
- What effect does pH have on the rate of enzyme activity? Explain this with respect to the nature of enzymes.
- Predict the optimal pH for turnip peroxidase activity.
- What influence do you think a cold temperature (ice bucket or 0-4ºC) would have on the rate of the turnip peroxidase activity?
- What influence do you think a warm temperature (around human body temperature or fever 37-40ºC) would have on the rate of turnip peroxidase activity?
- What outcome can you predict of the rates when changing the concentration of enzyme or substrate?
Set-up and Calibrate SpectroVis Plus
- Plug SpectroVis to computer via USB cable
- Launch Vernier Spectral Analysis (https://www.vernier.com/product/spectral-analysis/)
- Begin a new experiment by choosing “Absorbance vs. Time (Kinetics)”
- Wait for Lamp to warm up if necessary
- Place a blank cuvette in the unit and press “Finish Calibration”
- Enter a wavelength of 500nm and press “done”
- Proceed to the individual exercises
Effect of pH on Peroxidase Activity
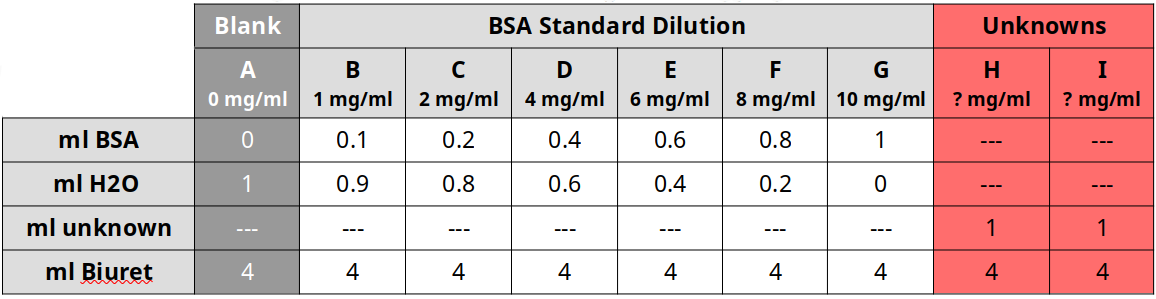
- Set-up cuvette for pH 3
- Add 10 drops 0.02% hydrogen peroxide
- Add 5 drops 0.2% guaiacol
- Add 20 drops of pH 3 buffer
- Add 10 drops turnip extract (enzyme) last
- Quickly invert the cuvette to mix and place the upright cuvette into the Spectrovis Plus
- Press “Collect” to begin recording data
-
- Stop data collection after 200s
- “Export Data” as a “.csv” file to computer or a USB flash drive
- Ensure that it has saved as a “.csv” file
- this will open in a text editor or spreadsheet
-
- Sequentially repeat the experiment exchanging the pH buffer with pH 5, 7, 10

Effect of Temperature on Peroxidase Activity
- Buffers are incubated on ice (0ºC), at room temperature (20ºC), in 40ºC or 60ºC at least 5 minutes
- Set-up cuvette for 0ºC
- Add 10 drops 0.02% hydrogen peroxide (incubated at 0ºC for 10 minutes)
- Add 5 drops 0.2% guaiacol
- Add 20 drops of extraction buffer at 0ºC
- Add 10 drops turnip extraction (incubated at 0ºC for 10 minutes) last
- Quickly invert the cuvette to mix and place the upright cuvette into the Spectrovis Plus
- Press “Collect” to begin recording data
- Stop data collection after 200s
- “Export Data” as a “.csv” file to computer or a USB flash drive
- Ensure that it has saved as a “.csv” file
- this will open in a text editor or spreadsheet
- Sequentially repeat the experiment exchanging the buffers stored at 20ºC, 40ºC, 60ºC
Effect of Substrate Concentration on Peroxidase Activity
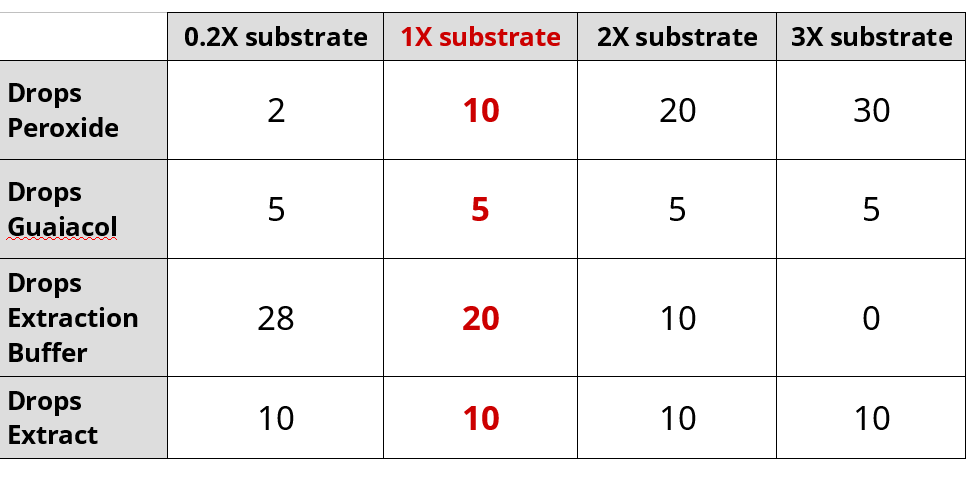
- Set-up cuvette for 1X substrate
- Add 10 drops 0.02% hydrogen peroxide
- Add 5 drops 0.2% guaiacol
- Add 20 drops of extraction buffer
- Add 10 drops turnip extraction last
- Quickly invert the cuvette to mix and place the upright cuvette into the Spectrovis Plus
- Press “Collect” to begin recording data
-
- Stop data collection after 200s
- “Export Data” as a “.csv” file to computer or a USB flash drive
- Ensure that it has saved as a “.csv” file
- this will open in a text editor or spreadsheet
-
- Sequentially repeat the experiment with differing amounts of buffer and peroxide:
-
- Repeat the experiment with 0.2X substrate and export data to USB drive
- Repeat the experiment with 2X substrate and export data to USB drive
- Repeat the experiment with 3X substrate and export data to USB drive
-
Effect of Enzyme Concentration on Peroxidase Activity
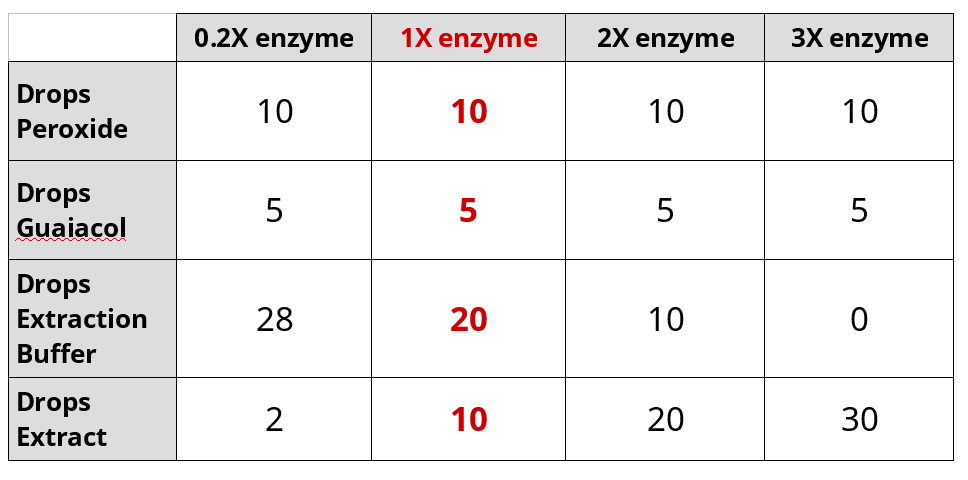
- Set-up cuvette for 1X enzyme
- Add 10 drops 0.02% hydrogen peroxide
- Add 5 drops 0.2% guaiacol
- Add 20 drops of extraction buffer
- Add 10 drops turnip extraction last
- Quickly invert the cuvette to mix and place the upright cuvette into the Spectrovis Plus
- Press “Collect” to begin recording data
-
- Stop data collection after 200s
- “Export Data” as a “.csv” file to computer or a USB flash drive
- Ensure that it has saved as a “.csv” file
- this will open in a text editor or spreadsheet
-
- Sequentially repeat the experiment with differing amounts of buffer and extract:
-
- Perform the experiment on 1X enzyme and export data to USB drive
- Repeat the experiment with 0.2X enzyme and export data to USB drive
- Repeat the experiment with 2X enzyme and export data to USB drive
- Repeat the experiment with 3X enzyme and export data to USB drive
-
Tags: quantitative reasoning














") and
and ")

 .
.
 and
and 

 = \sqrt{\sum_{k=1}^{p}(X_{ik} - X_{jk})^2}")






 [GFDL (http://www.gnu.org/copyleft/fdl.html) or CC-BY-SA-3.0 (http://creativecommons.org/licenses/by-sa/3.0/)], via Wikimedia Commons")



