The Hardy-Weinberg principle is a mathematical model used to describe the equilibrium of two alleles in a population in the absence of evolutionary forces. This model was derived independently by G.H. Hardy and Wilhelm Weinberg. It states that the allele and genotype frequencies across a population will remain constant across generations in the absence of evolutionary forces. This equilibrium makes several assumptions in order to be true:
An infinitely large population size
The organism involved is diploid
The organism only reproduces sexually
There are no overlapping generations
Mating is random
Allele frequencies equal in both genders
Absence of migration, mutation or selection
As we can see, many items in the list above can not be controlled for but it allows for us to make a comparison in situations where expected evolutionary forces come into play (selection etc.).
Hardy-Weinberg Equilibrium
The alleles in the equation are defined as the following:
Genotype frequency is calculated by the following:
Allele frequency is calculated by the following:
In a two allele system with dominant/recessive, we designate the frequency of one as p and the other as q and standardize to:
Therefore the total frequency of allalleles in this system equal 100% (or 1)
Likewise, the total frequency of all genotypes is expressed by the following quadratic where it also equals 1:
This equation is the Hardy-Weinberg theorem that states that there are no evolutionary forces at play that are altering the gene frequencies.
Calculating Hardy-Weinberg Equilibrium (activity)
This exercise refers to the PTC tasting exercise. One can test for selection for one allele within the population using this example. Though the class size is small, pooling results from multiple section can enhance the exercise. Remember to surmise the dominant/recessive traits from the class counts.
What is the recessive phenotype and how can we represent the genotype?
What is the dominant phenotype and how can we represent the genotypes?
What is the frequency of recessive genotype? (q2)
What is the frequency of the recessive allele? (q)
What is the frequency of the dominant allele?(p=1-q)
Use Hardy-Weinberg to calculate the frequency of heterozygotes in the class. (2pq)
Use Hardy-Weinberg to calculate the frequency of homozygotes in the class. (p2)
Using an aggregate of multiple section, compare the local allelic and genotypic frequencies with what the Hardy-Weinberg would predict.
With this small number in mind, we can see that there are problems with the assumptions required for this principle. The instructor will perform the following simulation in class to illustrate the effects on multiple populations with the effects of selection and /or population limitations. A coefficient of fitness can be applied to illustrate a selective pressure against an allele.
In the case of a selective pressure, a fitness coefficient (w) can be introduced. A research article http://www.jci.org/articles/view/64240 has shown that the Tas2R38 receptor aids in the immune response against Pseudomonas. Imagine a situation where there is an epidemic of antibiotic resistant Pseudomonas. This would
show that the dominant allele will have a selective advantage.
Modify the fitness coefficient in the Population Genetics Simulator and describe the effects this would have over many successive generations.
A case study of evolution: Population Genetics at Work
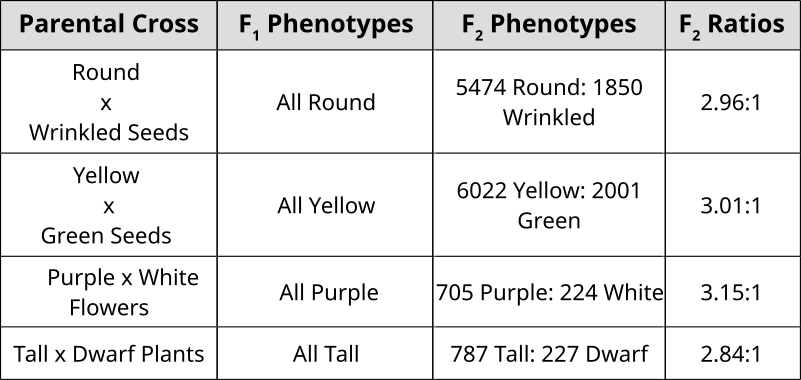
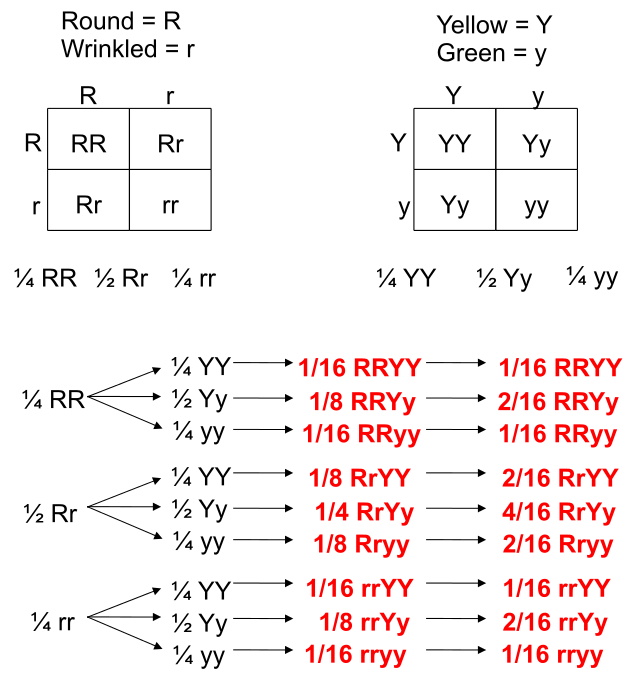
Punnett Squares are convenient for predicting the outcome of monhybrid or dihybrid crosses. The expectation of two heterozygous parents is 3:1 in a single trait cross or 9:3:3:1 in a two-trait cross. Performing a three or four trait cross becomes very messy. In these instances, it is better to follow the rules of probability. Probability is the chance that and event will occur expressed as a fraction or percentage. In the case of a monohybrid cross, 3:1 ratio means that there is a 3/4 (0.75) chance of the dominant phenotype with a 1/4 (0.25) chance of a recessive phenotype.
A single die has a 1 in 6 chance of being a specific value. In this case, there is a 1/6 probability of rolling a 3. It is understood that rolling a second die simultaneously is not influenced by the first and is therefore independent. This second die also has a 1/6 chance of being a 3.
We can understand these rules of probability by applying them to the dihybrid cross and realizing we come to the same outcome as the 2 monohybrid Punnett Squares as with the single dihybrid Punnett Square.
This forked line method of calculating probability of offspring with various genotypes and phenotypes can be scaled and applied to more characteristics.
Measurements can be made of individual genes of interest through PCR of those specific genes. A process known as Real-Time PCR or quantitative PCR (qPCR) is used to measure individual genes using fluorescence measurements. An intercalating agent that binds only to double stranded DNA called Sybr Green is used in a qPCR machine that is measuring fluorescence after each cycle of PCR indirectly indicates the amount of amplified product. However, non-specific products of amplification may also be measured and not discriminated from the authentic amplicon.
An alternative to Sybr Green is exemplified by the TaqMan technology. With TaqMan, a third primer (TaqMan probe) is designed in the middle of the area to be amplified. This middle primer is designed with a hairpin self-complimentarity so that the 5′ and 3′ ends are in close proximity. At one end, a fluorescent reporter is attached while the other terminus has a quencher that absorbs any fluorescence signal. Under normal circumstances, measurements of fluorescence will be very low. When PCR extension occurs, the Polymerase hydrolyzes this middle primer, thereby separating the quencher and reporter. The name TaqMan is a play on words since it is imagined that the polymerase is chewing up the probe like Pacman. With increased distance between quencher/reporter, fluorescence signal from this probe can now be measured. This method is much more specific than Sybr Green, however the use of specific probes increases the cost considerably.
Threshold Cycles (Ct)
Credit: Zuzanna K. Filutowska (CC-BY-SA 3.0)Fluorescence measurement early during the PCR process will be very low due to small number of dsDNA molecules (Sybr Green) or most TaqMan primers being quenched. During this exponential DNA production, a threshold will be reached in which the fluorescence will linearly increase. A specific point where the fluorescence is clearly measurable called the Threshold Cycle (Ct) is used as a reference point to compare expression values.
Looking at the example of Sybr Green qPCR above, it can be observed that samples exponentially increasing at a lower Cycle number (Ct) has a higher level of mRNA expression (towards the left) of that gene than samples with higher cycle number (towards the right). Notice that the fluorescence eventually plateaus and stops increasing. This is due to the depletion of raw materials for DNA production like dNTPs.
Since the PCR reactions theoretically represent a doubling of DNA after each cycle, the Ct values can be interpreted on a base 2 system. If there is a difference in Ct between two samples (ΔCt) of 5 cycles, this corresponds to 25 or 32 fold difference. We can control for variations in the RNA preparation through comparing the fluorescence values of our gene of interest to a housekeeping gene like actin. The use of a house-keeping gene to normalize the initial input to the reactions and comparison between samples is referred to as Relative Quantification.
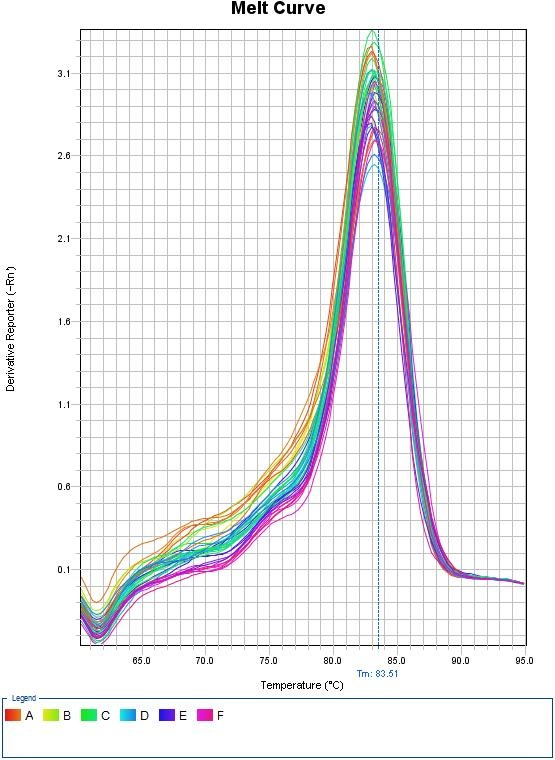
Melt Curves for Sybr Green
Top panel illustrates the decrease in fluorescence as the temperature increases due to the dissociation of double stranded DNA. Bottom panel illustrates the first derivative plot. Each peak in this example illustrates a different allele. The double peaks represent the presence of the 2 distinct alleles in the amplification products. Credit: Seans Potato Business (CC-BY-SA 3.0)When using Sybr Green, we need to ensure that the PCR is specific so that the fluorescence measurement truly reflect amplification of our gene of interest. At the end of each qPCR run (~40 cycles), a melt curve is performed. A melting curve (or dissociation curve) comes from constant measurements as the temperature is increased. As temperature increases, the DNA strands start to denature and fluorescence will begin to decrease. After complete separation of DNA strands, the fluorescence will again remain constant. The way this curve is viewed is through a derivative plot where the inflection in fluorescence reading is reported as the melting temperature (Tm).
This melt curve illustrates each sample contains the same specific product with a melting temperature of 83.51°C.
Any peaks in this plot refer to a specific PCR product. If multiple peaks appear, the results will not be valid as they do not directly measure a single product.
Expression measurements
Differential gene expression refers to transcriptional programs activated by the cell under various conditions. “Differential” refers to a comparison of two or more states or timepoints. Using mRNA as an indirect measurement of protein, one can ascertain which proteins are linked to these different states. In eukaryotes, this can be assessed by enriching total RNA for polyA-containing mature mRNA. Through the use of oligo-(dT) containing resin, mRNA can be separated from non-protein encoding RNA. Likewise, performing a reverse-transcription using an oligo-(dT) primer will create a stable complimentary DNA (cDNA) molecule that can be used with PCR. Using qPCR in this way is called RT-PCR or reverse-transcription polymerase chain reaction where specific primer pairs are used to amplify a small portion of a known gene.
Hybridization based methods and Microarrays
Credit: FrozenMan (CC-BY-SA 4.0)Prior to RT-PCR, expression of individual genes was assessed through a hybridization-based approach. This method called for running RNA on an agarose gel and transferring the size-fractionated RNAs onto a membrane through a method called “blotting”. This transferred RNA was then hybridized to a radioactively labelled probe for a specific gene (corresponding to the reverse complimentary sequence) and visualized by exposure to X-ray film in a process called Northern Blotting. The intensity of the band would be proportional to the amount of mRNA corresponding to the gene of interest. Re-probing with a housekeeping gene like actin would be used as a loading control to illustrate that a similar amount of total RNA was loaded into each well. Differences in sizes of the mRNA on the Northern Blot also revealed differences in splice variants of mature mRNA in the different states.
Credit: Jeremy Seto (CC-BY-NC-SA)
This technique was later adapted using non-radioactive methods. Using these non-radioactive methods, the reverse protocol was developed to measure multiple gene targets. By systematically immobilizing gene specific probes onto a membrane or a microscope slide, an array of targets can be produced. In the simplest paradigm of having 2 states (control or experimental), cDNA from each sample can be used to generate fluorescent RNA that can hybridize to immobilized probes. Using 2 different fluorescent markers allows for the competitive hybridization onto the array whereby the fluorescent signal in each channel can reveal the differential gene expression of the two states in a 2-color microarray.
Detection and measuring amount of a virus by RT-PCR
COVID-19 is a disease caused by SARS-CoV2 virus. This virus is characterized as a (+) ss-RNA virus with a genome of about 30kb.
In order to detect this virus, respiratory tract samples are collected and the RNA extracts are subjected to reverse transcription followed by PCR of a portion of the N gene. The exponential nature of PCR permits for the detection of the complementary DNA in minuscule concentrations. By using standards of known concentrations, quantitative PCR (qPCR) effectively determine the viral load within the cells.
The following video demonstrates the lab work involved in titrating Adeno-Associated Virus (AAV) by qPCR. While AAV has a genome of ssDNA, this video illustrates similar steps in quantitation.
Co-dominance is said to occur when there is an expression of two dominant alleles. The prototypical case for this is the human ABO blood grouping.
Three alleles exist in the ABO system: A, B and O. This results in four blood types: A, B, O and the blended AB.
Incomplete Dominance
During Mendel’s time, people believed in a concept of blending inheritance whereby offspring demonstrated intermediate phenotypes between those of the parental generation. This was refuted by Mendel’s pea experiments that illustrated a Law of Dominance. Despite this, non-Mendelian inheritance can be observed in sex-linkage and co-dominance where the expected ratios of phenotypes are not observed clearly. Incomplete dominance superficially resembles the idea of blending inheritance, but can still be explained using Mendel’s laws with modification. In this case, alleles do not exert full dominance and the offspring resemble a mixture of the two phenotypes.
Incomplete dominance in snapdragon flowers superficially appears like blending inheritance.Credit: Jeremy Seto (CC-BY-NC-SA) The most obvious case of a two allele system that exhibits incomplete dominance is in the snapdragon flower. The alleles that give rise to flower coloration (Red or White) both express and the heterozygous genotype yields pink flowers. There are different ways to denote this. In this case, the superscripts of R or W refer to the red or white alleles, respectively. Since no clear dominance is in effect, using a shared letter to denote the common trait with the superscripts (or subscripts) permit for a clearer denotation of the ultimate genotype to phenotype translations.
Problem: Incomplete Dominance
If pink flowers arose from blending inheritance, then subsequent crosses of pink flowers with either parental strain would continue to dilute the phenotype. Using a Punnet Square, perform a test cross between a heterozygous plant and a parental to predict the phenotypes of the offspring.
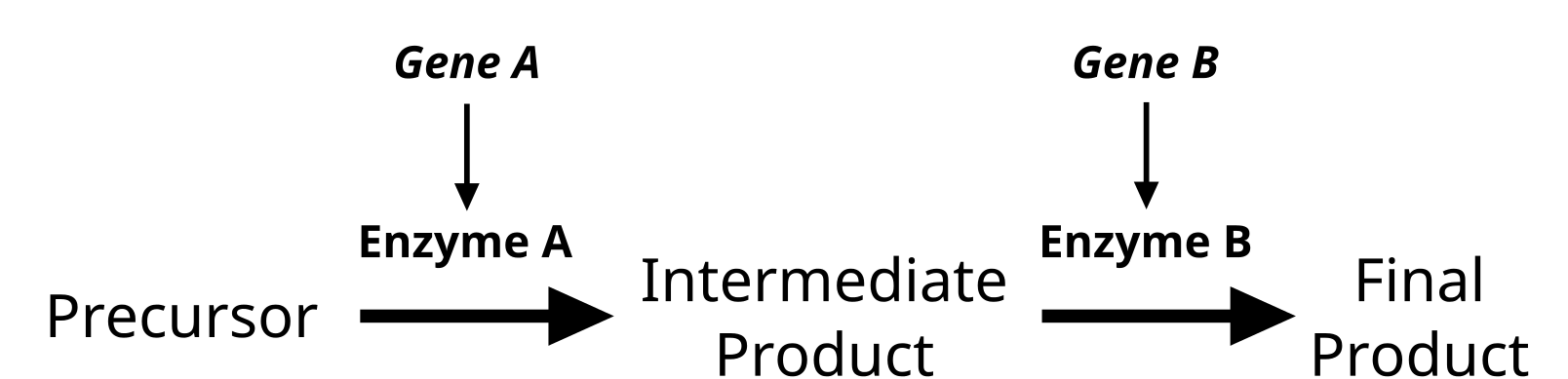
Epistasis and Modifier Genes
Interplay of multiple enzymes in a biochemical pathway will alter the phenotype. Some genes will modify the actions of another gene. Credit: Jeremy Seto (CC0)
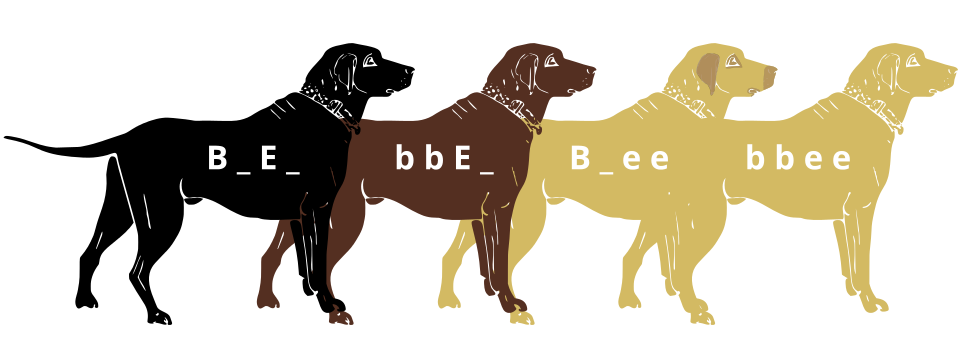
Genes do not exist in isolation and the gene products often interact in some way. Epistasis refers to the event where a gene at one locus is dependent on the expression of a gene at another genomic locus. Stated another way, one genetic locus acts as a modifier to another. This can be visualized easily in the case of labrador retriever coloration where three primary coat coloration schemes exist: black lab, chocolate lab and yellow lab.
Chocolate lab (top), Black lab (middle), Yellow lab (bottom) coat colorations arise from the interaction of 2 gene loci, each with 2 alleles. Credit: Erikeltic [ CC-BY-SA 3.0]
Two genes are involved in the coloration of labradors. The first is a gene for a protein called TYRP1, which is localized to the melanosomes (pigment storing organelles). Three mutant alleles of this gene have been identified that reduce the function of the protein and yield lighter coloration. These three alleles can be noted as “b” while the functioning allele is called “B“. A heterozygous (Bb) or a homozygous dominant individual will be black coated while a homozygous recessive (bb) individual will be brown.
The second gene is tied to the gene for Melanocortin 1 Receptor (MC1R) and influences if the eumelanin pigment is expressed in the fur. This gene has the alleles denoted “E” or “e“. A yellow labrador will have a genotype of either Bbee or bbee.
Black lab (EE or Ee) and Yellow lab (ee) [CC0] The interplay between these genes can be described by the following diagram:
For the most part, mammals have gender determined by the presence of the Y chromosome. This chromosome is gene poor and a specific area called sex determining region on Y (SRY) is responsible for the initiation of the male sex determination. The X-chromosome is rich in genes while the Y-chromosome is a gene desert. The presence of an X-chromosome is absolutely necessary to produce a viable life form and the default gender of mammals is traditionally female.
Human X and Y chromosomes with G-Banding.
Chromosomal painting techniques can reveal the gender origin of mammalian cells. By using fluorescent marker sequences that can hybridize specifically to X or Y chromosomes through Fluorescence In Situ Hybridization (FISH), gender can be identified in cells.
DNA can be cut by restriction endonucleases (RE). Endonucleases are enzymes that can hydrolyze the nucleic acid polymer by breaking the phosphodiester bond between the phosphate and the pentose on the nucleic acid backbone. This is a very strong covalent bond while the weaker hydrogen bonds maintain their interactions and double strandedness.
As the name implies, restriction endonucleases (or restriction enzymes) are “restricted” in their ability to cut or digest DNA. The restriction that is useful to biologists is usually palindromic DNA sequences. Palindromic sequences are the same sequence forwards and backwards. Some examples of palindromes: RACE CAR, CIVIC, A MAN A PLAN A CANAL PANAMA. With respect to DNA, there are 2 strands that run antiparallelel to each other. Therefore, the reverse complement of one strand is identical to the other. Molecular biologists also tend to use these special molecular scissors that recognize palindromes of 6 or 8. By using 6-cutters or 8-cutters, the sequences occur throughout large stretches rarely, but often enough to be of utility.
EcoRI generates sticky of cohesive endsSmaI generates blunt ends
Restriction enzymes hydrolyze covalent phosphodiester bonds of the DNA to leave either “sticky/cohesive” ends or “blunt” ends. This distinction in cutting is important because an EcoRI sticky end can be used to match up a piece of DNA cut with the same enzyme in order to glue or ligate them back together. While endonucleases cut DNA, ligases join them back together. DNA digested with EcoRI can be ligated back together with another piece of DNA digested with EcoRI, but not to a piece digested with SmaI. Another blunt cutter is EcoRV with a recognition sequence of GAT | ATC.
Restriction fragment length polymorphism (RFLP) is a technique that exploits variations in DNA sequences. DNA from differing sources will have variations or polymorphisms throughout the sequence. Using Restriction Enzymes, these differences in sequences may be teased out. However, if one were to take the entirety of the human genome and chop it up with a restriction enzyme, many indecipherable fragments would be made. In fact, the resulting agarose gel would simply show a large smear of DNA. RFLP analysis requires that a probe to a specific area of DNA be used to identify specific locations. Agarose gels would be transferred to a membrane or filter where they would be hybridized to these radioactive probes.
Homologous chromosomes with restriction sites noted by triangles. the rectangle sitting on the chromosomes correspond to a probe locus. Credit: Jeremy Seto (CC0)
RFLP analysis was designed for forensic science to discriminate between people. Since people are 2N, they have pairs of homologous chromosomes with the same loci. However, these loci may contain different alleles. In this case, the phenotype for these alleles is the actual sequence that may or may not contain restriction sites. The presence or absence of a restriction site may arise from single nucleotide polymorphisms (SNPs) that reveal the natural variation between people.
The schematic below illustrates a comparison of restriction profiles between two sources. Note that the probe overlaps a restriction site in one of the alleles. This probe will be able to bind to both fragments given sufficient sequence overlap. Upon resolving on an agarose gel, genomic DNA that does not hybridize with the probe will obscure the locus of interest as a large smear. A filter is placed on top of the agarose and pressed against it to transfer the DNA in a process called Southern Blotting. Following a lengthy transfer, the filter is denatured to and incubated with the radioactive probe. To visualize this probe hybridization, film is exposed to the filter and processed.
Following restriction digestion, the samples are resolved on an agarose gel. Digestion of genomic DNA will result in a large smear. Following transfer of the DNA onto a membrane through capillary action, the membrane is probed with radioactive probe DNA. Probe binds selectively to complementary sequences to reveal a series of distinct bands. An interactive demonstration of the first DNA fingerprinting. Credit: Oder Zeichner: abigail [ or CC-BY-SA-3.0] / Autoradiogram
Sample A only reveals one band after processing because this person is homologous for the same allele. Sample B is heterozygous and reveals three bands. Credit: Retama (CC-BY-SA 4.0) RFLPs represent inheritable markers and can reveal relationships between different individuals. A pedigree can illustrate the relationship of the inherited alleles. The technique can be more informative if using multiple probes simultaneously for different loci or to use multi-locus probes that hybridize to multiple locations. A crude simulation of the procedures for RFLP analysis can be found at https://stories.wgbh.org/create-dna-fingerprint/.
RFLPs may arise from differences in the STR/VNTR repeats between restriction sites. Credit: Jeremy Seto (CC0)
While RFLPs can arise from SNPs, they may also be caused by the expansion or contraction of repeated elements between restriction sites. These repeated elements of DNA are referred to as Variable Number Tandem Repeats (VNTR) and illustrate polymorphisms that normally occur in non-coding regions of the genome.
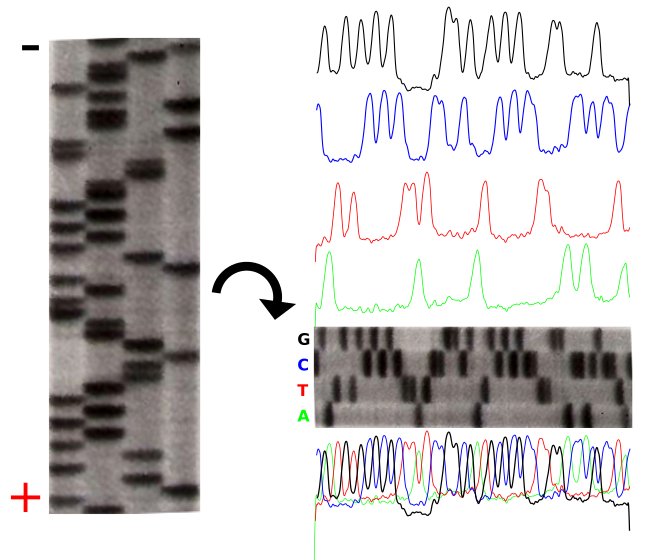
The polymerization of nucleic acids occurs in a 5′ → 3′ direction. The 5′ position has a phosphate group while the 3′ position of the hexose has a hydroxyl group. Polymerization depends on these 2 functional groups in order for a dehydration synthesis reaction to occur and extend the sugar-phosphate backbone of the nucleic acid. In the 1970’s, Fred Sanger’s group discovered a fundamentally new method of ‘reading’ the linear DNA sequence using special bases called chain terminators or dideoxynucleotides. The absence of a hydroxyl group at the 3′ position blocks the polymerization resulting in a termination. This method is still in use today it is called: Sanger dideoxynucleotide chain-termination method. This method originally used a radioactively labeled primer to initiate the sequencing reaction. Four reactions take place where each reaction is intentionally “poisoned” with a dideoxy chain terminator. For example, 1 reaction will have all 4 dNTPs (deoxynucleotide triphosphates)with the addition to a small amount of ddATP (dideoxyadenosine triphosphate). This reaction will result in a series of premature terminations of the polymerization specifically at different locations where an Adenine would be incorporated.
dATP is a natural monomer used in the polymerization of DNA. The 3′-OH is the attachment point of the next subsequent nucleotide.The lack of a 3′-OH in this molecule of ddATP makes it a chain terminator that will prohibit the addition of another nucleotide to the DNA polymerThe product of these 4 separate sequencing reactions is run on a large polyacrylamide sequencing gels. The smallest fragments run through the gel the fastest and create a ladder-like pattern. This can be visualized through use of an x-ray film that is sensitive to the radioactivity. Each lane of the gel corresponds to one of the four chain terminating reactions. The bases are read sequentially from the bottom up and reveals the sequence of the DNA.
The sequencing gel can be manually scored. The profiles of each lane have been created using ImageJ to illustrate the banding pattern and subsequent sequence. Credit: John Schmidt& Jeremy Seto(CC-BY-SA 3.0)
Fluorescent Chain Termination and Capillary Electrophoresis
Credit: Estevezj (CC-BY-SA 3.0)Radioactivity is dangerous and undesirable to work with so chain terminators with fluorescent tags were developed. This method synthesizes a series of DNA strands that are specifically fluorescent at the termination that is passed through a capillary electrophoresis system. As the fragments of DNA pass a laser and detector, the different fluorescent signal attributed to each ddNTP is identified and generates a chromatogram to represent the sequence. Fluorescent Chain Terminators are now used in reactions and run through a small capillary. The smallest fragments run through first and are detected to reveal a chromatogram.
Fluorescent Chromatograms are used to score the the nucleotide chain termination. The amplitude of each peak corresponds to the strength or certainty of the nucleotide call. Chromatogram files are usually provided alongside the sequence file with the extension *.ab1 while the sequence files are provided as a text file in the fasta format. More about these files can be found here. The ab1 files are extremely important to analyze when there is ambiguity or sequencing errors. These ab1 files can also be used to ascribe a quality score on the base call.
When there is too much ambiguity in the signal because of multiple peaks, you will often find a N in place of one of the 4 nucleotides (A,T,C,G).
Sequencing Genomes
Credit:Jeremy Seto (CC-BY-NC-SA 3.0)
Traditional sequencing of genomes was a long and tedious process that cloned fragments of genomic DNA into plasmids to generate a genomic DNA library (gDNA). These plasmids were individually sequenced using Sanger sequencing methodology and computational was performed to identify overlapping pieces, like a jigsaw puzzle. This assembly would result in a draft scaffold.
The video below is taken from yourgenome.org (CC-BY) and illustrates the sequencing of the human genome through the shotgun sequencing approach.
RNA purification occurs similar to DNA preparations. A silica based column is used where DNA is excluded from binding based on size and through an additional DNA digestion step using the enzyme DNase I. RNA is extremely fragile and prone to degradation. Because of this, separate pipettes and plastics are usually used in labs to reduce the amount of exposure to environmental or experimental RNase. When handling RNA, be extremely careful of contaminating the buffers or samples. Always wear gloves as skin carries RNase enzymes. Refrain from talking as to not contaminate the area with RNase found in saliva.
RLT = RNA lysis buffer: contains guanidine, a harsh denaturant RW1 = RNA wash buffer RPE = Second RNA wash buffer with Ethanol RDD = DNase digestion buffer
Harvest a maximum of 1 x 107 cells, as a cell pellet or by direct lysis in the vessel. Add the appropriate volume of Buffer RLT and vortex vigorously.
If < 5 x 106 cells → 350 μl RLT (< 6cm plate)
if ≤ 1 x 107 cells → 600 μl RLT (6-10cm plate)
Add 1 volume of 70% ethanol to the lysate, and mix well by pipetting. Do not centrifuge. Proceed immediately to next step.
Transfer up to 700 μl of the sample, including any precipitate, to an spin column placed in a 2 ml collection tube (supplied).
Close the lid, and centrifuge for 15 s at ≥8000 x g.
Discard the flow-through.
Wash: Add 350 μl Buffer RW1 to spin column, close lid, centrifuge for 15 s at ≥8000 x g (≥10,000 rpm). Discard flow-through.
Add 10 μl DNase I stock solution (see above) to 70 μl Buffer RDD. Mix by gently inverting the tube
Remove DNA (optional): Add DNase I incubation mix (70 μl) directly to spin column membrane, and place on benchtop (20–30°C) for 15 min.
Wash: Add 350 μl Buffer RW1 to spin column, close lid, centrifuge for 15 s at ≥8000 x g. Discard flow-through.
Add 700 μl Buffer RW1 to the spin column. Close the lid, and centrifuge for 15 s at ≥8000 x g. Discard the flow-through.
Add 500 μl Buffer RPE to the spin column. Close the lid, and centrifuge for 15 s at ≥8000 x g. Discard the flow-through.
Add 500 μl Buffer RPE to the spin column. Close the lid, and centrifuge for 2 min at ≥8000 x g.
Discard all flow-through and centrifuge at full speed for 1 min to dry the membrane.
Place the spin column in a new 1.5 ml collection tube. Add 30 μl RNase-free water directly to the spin column membrane.
Close the lid, and centrifuge for 1 min at ≥8000 x g to elute the RNA.
Add 30 μl RNase-free water directly to the spin column membrane. Close the lid, and centrifuge for 1 min at ≥8000 x g to elute the RNA.
Reverse Transcription
The Central Dogma of Molecular Biology was proposed by Francis Crick, the co-describer of the double stranded helical structure of DNA. This “dogma” was a statement to describe the flow of genetic information to show that DNA houses or stores data that is transcribed into RNA that is subsequently translated from nucleotides into amino acids through the machinery of the ribosomes. Since DNA is relatively static in it’s ability to store genetic information, the expression of this stored data into the intermediate RNA or to the final protein product is of great significance. Imagine that the DNA in the nucleus of your cheek cells is identical to the DNA of the nucleus of cells in your liver. While the instructions are identical, these are clearly different cells that have a difference in expression of proteins. Imagine a hard drive on a computer that stores information as 1’s and 0’s. These 1’s and 0’s do not have meaning until specific programs are called upon to act on this information. Likewise, different programs are called to use the instructions of your DNA to make a cheek cell different than a liver cell.
Credit: Daniel Horspool (CC-BY-SA 3.0)In 1970, Howard Temin and David Baltimore independently isolated an enzyme from the Rous Sarcoma Virus and Murine Leukemia Virus, respectively. This enzyme was capable of violating the Central Dogma. The genomes of these viruses consist of RNA, not DNA. During the infection process, this enzyme is responsible converting the RNA into DNA in a process called reverse transcription. This enzyme is logically called reverse transcriptase (RT). This discovery was rewarded with Nobel Prize in 1975. Later on, more viruses were discovered that were composed of RNA genomes that utilized this process, including HIV. Other enzymes within cells were also recognized to have reverse transcriptase activity, such as telomerase and retrotransposases. In molecular biology, these enzymes are used to convert mRNA into complementary copies of DNA called cDNA. The sum total of everything that is transcribed into RNA is referred to as the transcriptome. Synthesis of cDNA from any transcribed RNA can then be used for transcriptome analysis.
Exercise: Reverse Transcription of Eukaryotic mRNA
mRNA from eukaryotes are modified with 3′ polyadenylated tails. Oligo-dT primers can be used to prime the reverse transcription process of all mRNAs. All solutions should be kept on ice.
Determine the concentration of total RNA
Adjust concentration of RNA to 0.1mg/ml using Rnase free water
Combine 10 ml RNA, 1 ml Oligo-dT (50μM), and 1 ml dNTP Mix (10 mM each)
Denature mixture at 65°C for 5 minutes and then place on ice
Combine the following in a separate tube
4μl Buffer 5X → contains all salts and pH buffer
2μl 0.1 M DTT → a reducing agent to mimic the cellular environment
1μl RNaseOUT (40U/μl) → an RnaseA inhibitor
1μl SuperScript III RT → the reverse transcriptase enzyme
After the denatured mixture has been sufficiently cooled, ad 8μl enzyme mixture
incubate 45°C for 1 hour
deactivate enzyme by incubating 75°C for 10 minutes
The Central Dogma in Eukaryotes. Genomic DNA of genes often contain introns that are spliced out when an RNA matures to a mRNA. This excision of introns can result in splice variants of the same gene with variants of the same protein. Credit: Thomas Shafee(CC-BY 4.0)
Unlike prokaryotic genes, expression of genes in eukaryotic cells have complex systems of transcription factors that act on promoters to recruit RNA polymerases. Additionally, enhancer elements may reside many kilobase upstream of the promoter. These enhancers strengthen the transcription of the gene. In this case, transcription activator proteins or trans-activators augment the promoter activity.
1. DNA 2. Enhancer3. Promoter4. Gene5. Transcription Activator Protein6. Mediator Protein7. RNA Polymerase Credit: Jon Cheff (CC-BY-SA 4.0) Mediator proteins (coactivators) form a multiprotein complex with the activators to recruit RNA polymerase to the promoter.
Eukaryotic mRNA
Credit: Kelvinsong (CC-BY-3.0)
Eukaryotic genes may often contain introns(non-coding sequences) that are spliced out from the exons (coding sequences). This complexity permits for increased variety of gene products. Mature eukaryotic mRNAs conatins a 5′-methyl-Guanine followed by an untranslated leader sequence (5′-UTR), the coding sequences (cds), a 3′-untranslated region (3′-UTR) and a long stretch of Adenines (polyA tail).
Expression is most easily measured with RNA since nucleic acid manipulation is fairly simple with 4 different nucleotides. In eukaryotes, the messenger RNA (mRNA) intermediate that is transcribed from DNA contains a polyA tail that is used to separate these messages from other types of RNA that are abundant within cells (like ribosomal RNA). Through the use of an enzyme called reverse transcriptase (RT) and primers composed of deoxy-Thymidines (oligo-dT or dT18), mRNA can be converted into a single strand of DNA that is complimentary to the mRNA. This complimentary DNA is called cDNA. cDNA is very stable compared to the highly labile mRNA and is used for subsequent processing.





























 [GFDL (http://www.gnu.org/copyleft/fdl.html) or CC-BY-SA-3.0 (http://creativecommons.org/licenses/by-sa/3.0/)], via Wikimedia Commons")












.svg)




![Erikeltic [ CC-BY-SA 3.0]](https://commons.wikimedia.org/wiki/File:3labradorcols.jpg){kind=link}
![dmealiffe[CC BY-SA 2.0]](https://commons.wikimedia.org/wiki/File:Labrador_Retrievers_blackandchocolate.jpg){kind=link}
{kind=link}
![Janice Y Ahn, Jeannie T Lee [CC BY 2.0]](https://commons.wikimedia.org/wiki/File:X_Y_chromosome.jpg){kind=link}
![Oder Zeichner: abigail [ or CC-BY-SA-3.0]](https://commons.wikimedia.org/wiki/File:Southern-Blot-Agarosegel.jpg){kind=link}
{kind=link}
{kind=link}