Contents
Using UGENE to explore FASTA and GenBank files
Exploring Genbank Files (Activity)
- Download the file LacZ.gb and open in a text editor.
- This is a Genbank format file that contains the sequence following the word ‘ORIGIN‘ and terminating with ‘//‘.
- Prior to the sequence is a batch of descriptive information including references, organism and database cross-reference identifiers. While these don’t mean much to you, the appropriate database within Genbank can be queried to reveal more information about the sequence.
- Download the file LacZ.fasta and open in a text editor (NotePad).
- Notice the simple structure of the fasta file beginning with the ‘>’ and description of the sequence.
- This is a DNA sequence. But DNA is usually double stranded! We can assume the sequence of the second strand because it will be complimentary to this one.
- By convention: we know that this sequence is 5′ → 3′
- This text contains a portion of the E. coli genome that includes a gene called LacZ.
- This file does not contain any annotation to indicate where the gene sequence actually begins or ends.
- Launch UGENE and open both files. They will appear on the left side “Objects” pane.
- The default display automatically shows the reverse compliment of the DNA strand and all 6 Open Reading Frames (ORFs).
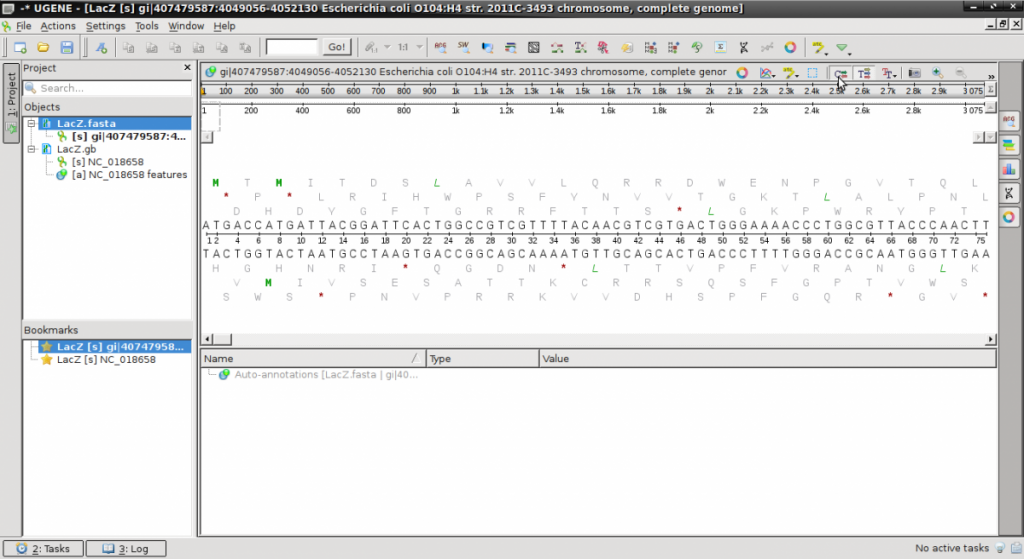
- To simplify the view, click on the ‘C‘ to remove the complimentary strand (look at the cursor in the image)
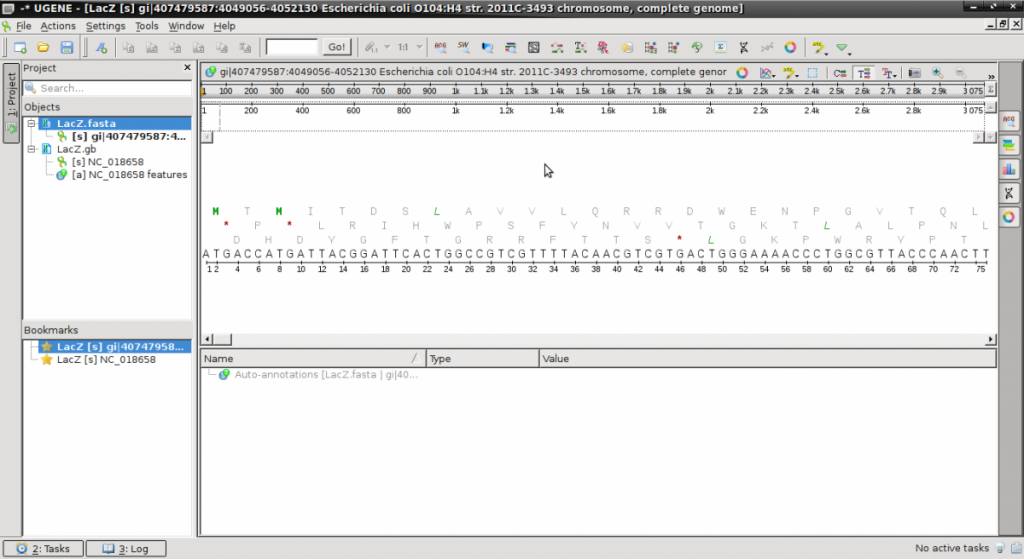
- The default display automatically shows the reverse compliment of the DNA strand and all 6 Open Reading Frames (ORFs).
- Count the ORFs :
- Find ORFs by right-clicking on the sequence and select “Analyze → Find ORFs“
- Default setting looks for ORFs on both strands with a minimum length of 100 nucleotides
- The Open Reading Frame here is defined as something beginning with initiation or start codons from the Standard Genetic Code (ATG) and two additional alternative start codons (TTG & CTG) that is terminated by any one of the three standard stop codons (TAA, TAG, TGA)
- Selecting Preview will provide the amount of possible ORFs fitting these criteria.
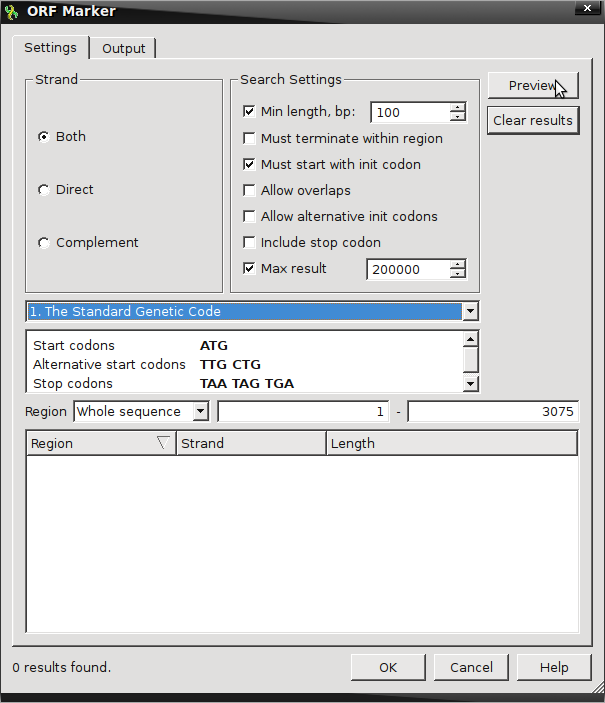
- Double click on the LacZ.gb in the Objects panel to activate the view.
- This file now shows the same sequence with information about the DNA

- Expand the various features in the Annotations pane at the bottom to explore the sequence features.
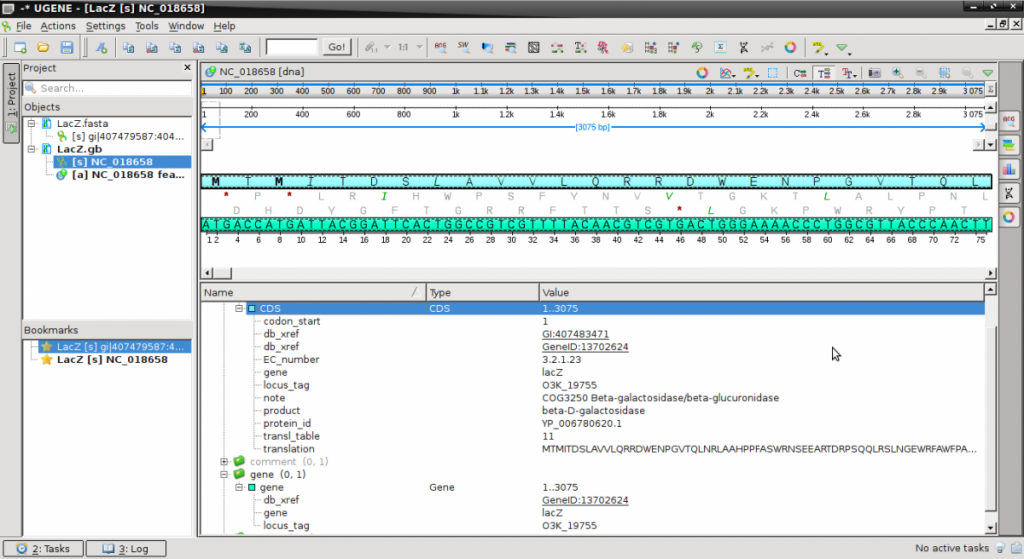
- This file now shows the same sequence with information about the DNA
Searching for Sequences
This video goes through the steps of finding sequences relevant to a study.

