Sequence output
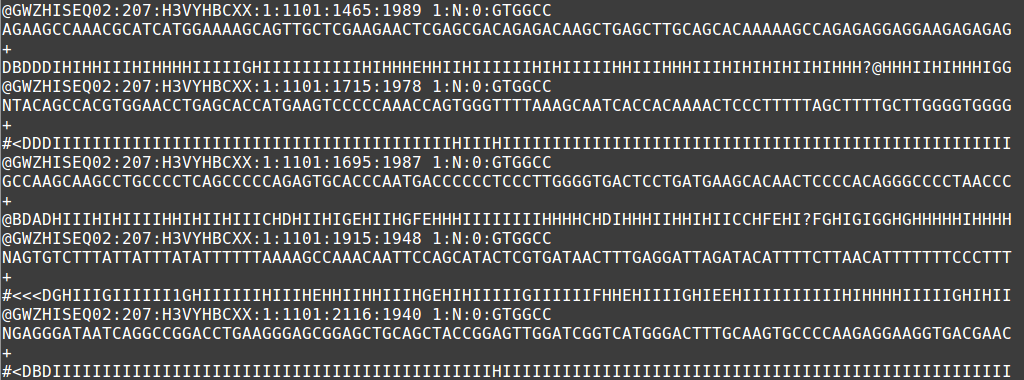
The output file of next generation sequencing methods utilize the fastq format. Like a fasta file, there is a header that describes the sequence. The first line is the header or title line which begins with ‘@‘ (remember that fasta begins with ‘>‘). The second line is the actual raw sequence (once again similar to fasta). The third line has no meaning while the fourth line is filled with symbols as long as the sequence line. This last line is the quality score of the base call. As with the Sanger sequencing, there may be ambiguity with the base call of the sequence and the certainty is maintained in the quality score.
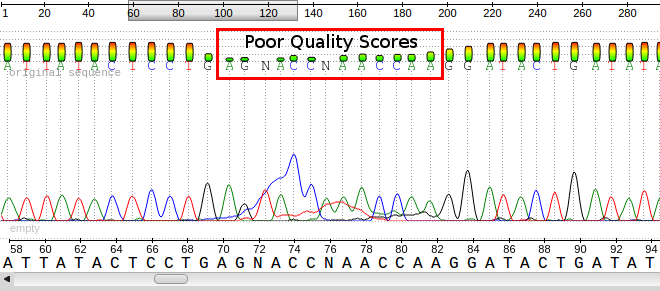
Phred scores were developed to assess the quality of the base calls arising from fluorescent Sanger sequencing during the Human Genome Project. The phred program scans the peaks of the chromatogram and scores based on certainty or accuracy of the call. The scores are logarithmically based and scores greater than 20 represent greater than 99% accuracy of the base call.
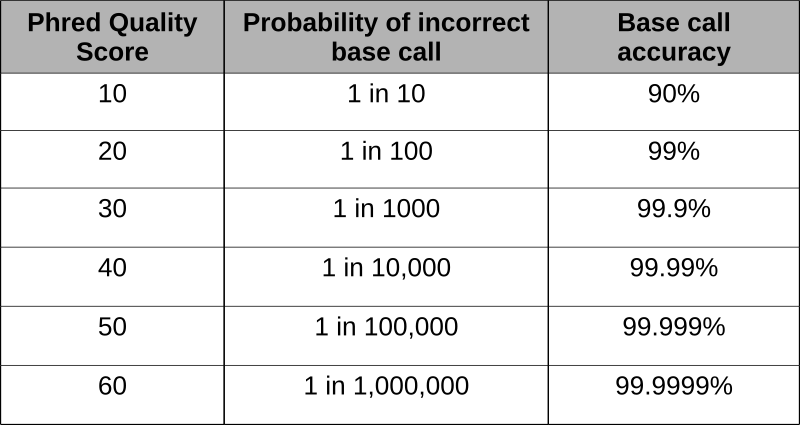
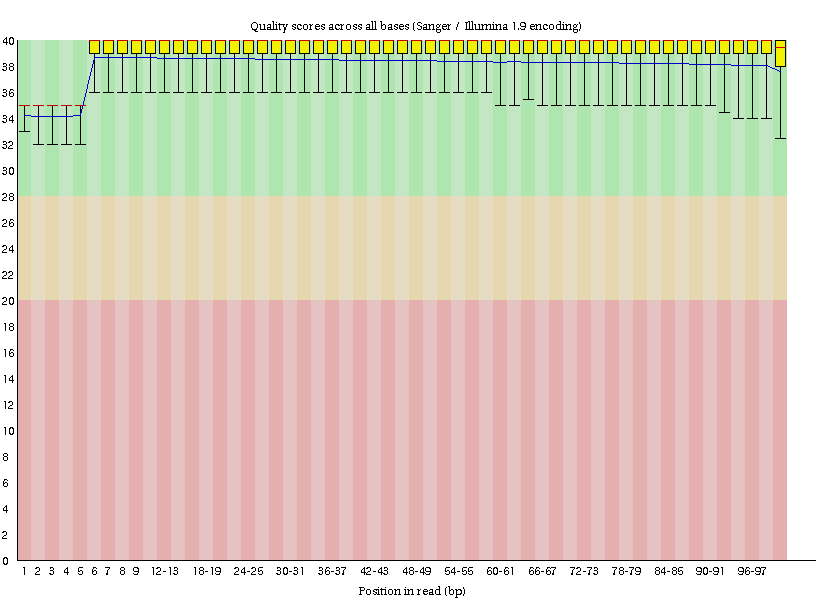
Using the phred scores embedded in the last line of fastq files, poor quality reads can be removed. Using a program like FastQC permits the assessment of the reads and produces graphical representation of quality.
Assembly and Alignment
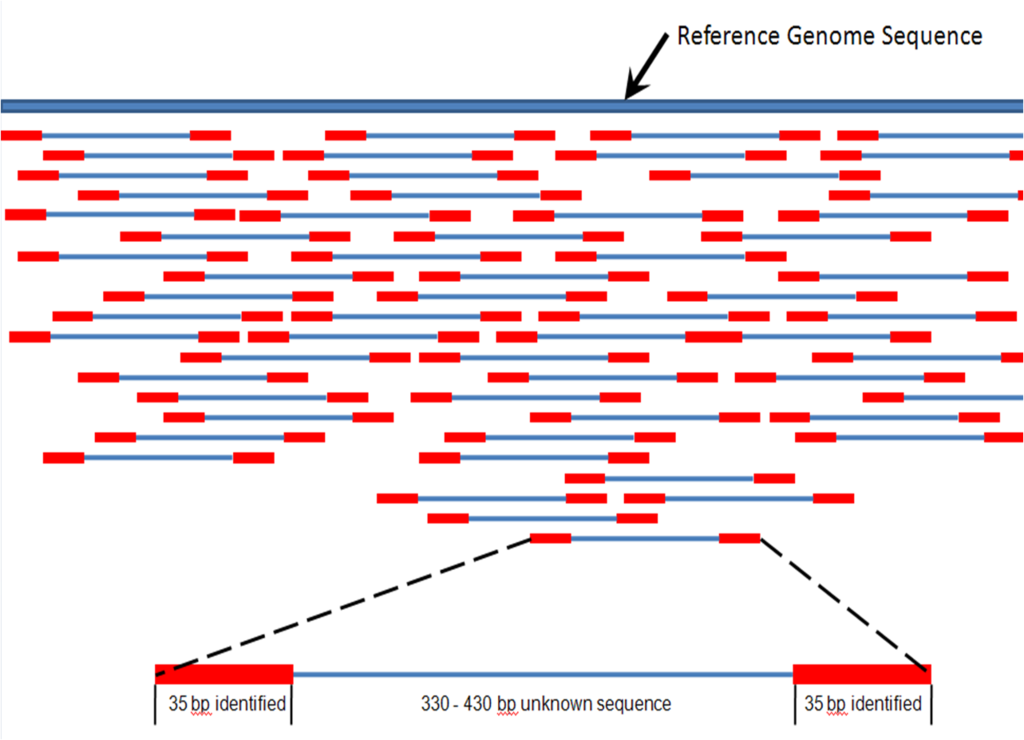
Sequences from short reads must be assembled into a usable sequence. To do so, a reference genome may aid in the assembly after adapter sequences are trimmed using automated methods. In the case that there is no reference genome, a related species may be used or a more computationally intensive process of de novo assembly must take place. With de novo assembly, it may be useful to have some long reads performed with PacBio to create scaffolds for generating the assembly into contiguous sequences, or contigs.