
Dr. Christopher Blair
BIO 4250
Molecular Evolution & Phylogenetics
The primary objectives of this lab are to introduce students to three important topics in phylogenetics including (1) databases; (2) multiple sequence alignment; (3) common file formats. Data that are used for phylogenetic analysis generally come from one or two sources. First, data can be collected de novo (e.g. sequencing new genes or collecting a suite of morphological characters) or data can be pulled from online repositories. One of the most popular databases is GenBank from the National Center for Biotechnology Information (NCBI). NCBI also maintains the composite database ENTREZ that allows users to query multiple databases simultaneously. Note that these databases are updated regularly, and the number of hits you see is likely to differ from those in the text.
Table of Contents
Databases
For phylogenetics, there are a few particularly important databases to be aware of. Navigate to the following website (ncbi.nlm.nih.gov/search/). One of the most important databases is the Nucleotide database under the Genomes heading (Fig. 1).

Fig. 1. Where to find the Nucleotide database on NCBI.
Clicking on Nucleotide will open a new link where you can specifically query a nucleotide database only. Two other databases of relevance are the Protein database and all of the literature databases that can be used to search for papers relevant to a specific subject or query.
Go ahead and search for human papillomavirus type 13 in the search bar and click Search. Again, this will search all the listed databases for human papillomavirus type 13 and display the results. Spend some time exploring the different databases to become familiar with them. For example, clicking on the PubMed link will eventually bring you to abstracts of relevant papers. Click on the Nucleotide link to bring up the following page (Fig. 2).
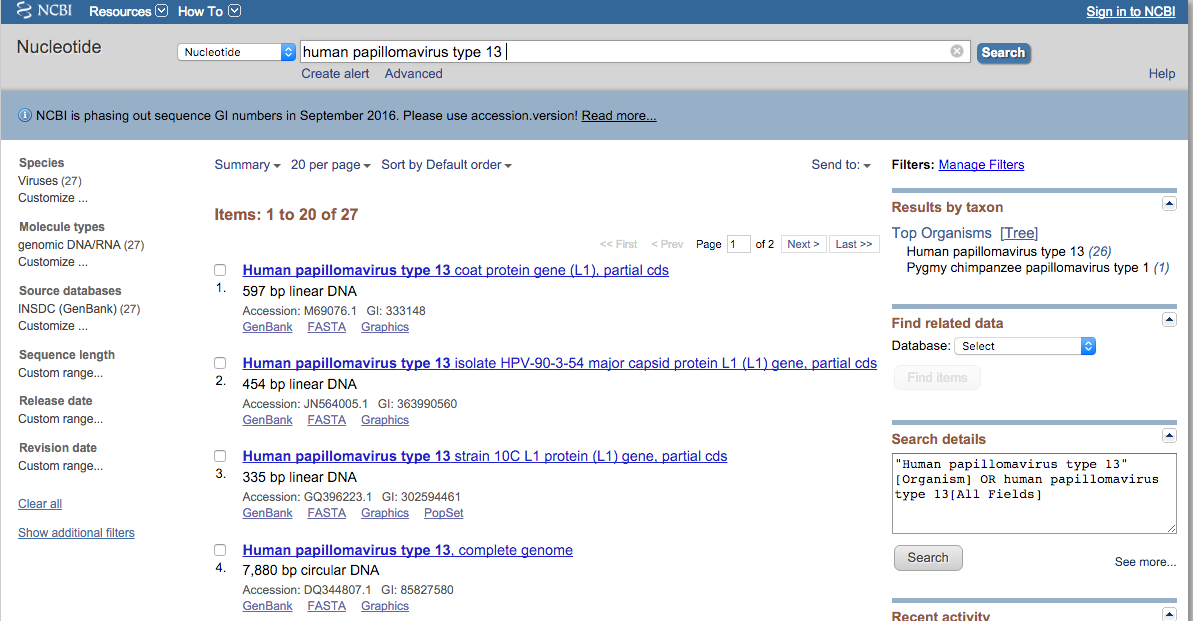
Fig. 2. Screenshot resulting from a search for human papillomavirus type 13 in the Nucleotide (GenBank) database.
This page lists nucleotide information from several genes pertaining to human papillomavirus type 13. Clicking on an item will bring up the full GenBank entry for the sequence (Fig. 3).
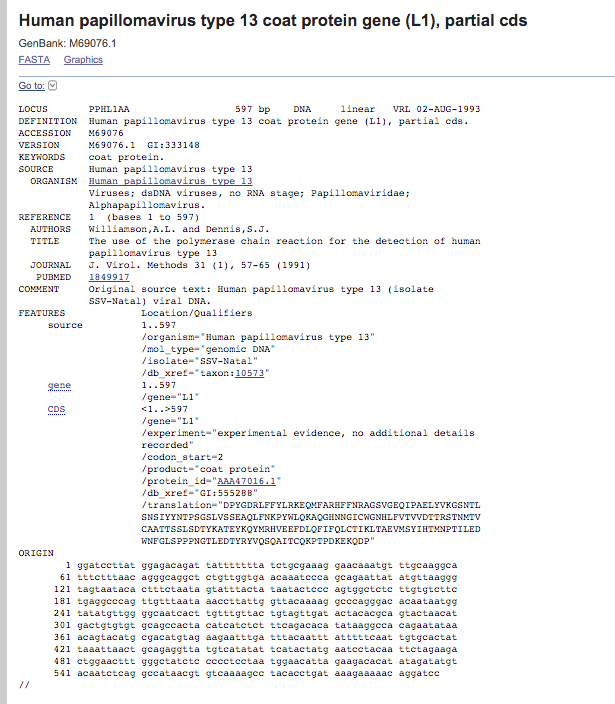
Fig. 3. Full GenBank entry for Accession Number M69076.1.
There is a lot of information presented in a GenBank entry that you should be familiar with. One of the most important items is the Accession Number, in this case M69076.1. This is the number that is used to identify a specific GenBank entry and is assigned by NCBI during sequence submission. You will also find other pieces of relevant information, including sequence length, annotation information, authors, title of article and journal of publication, etc. The term CDS refers to coding sequence and signifies that the sequence belongs to a protein coding gene. As such, GenBank provides both the full nucleotide sequence and the resulting translation in the correct reading frame.
Before we continue, it is a good idea to become familiar with some of the common file types used when working with sequence data. Probably the most common and one of the simplest is FASTA format. Each entry in a FASTA file begins with a > symbol followed by the taxon identifier and then the data.
Example of FASTA format:
>Species1
AATTGTCGAATTTGCTA
>Species2
AATTGCGTAATTGACTG
The vast majority of phylogenetic and sequence manipulation packages can work with FASTA files in some capacity. File extensions include .fasta, .fas.
A second file format to be familiar with is the NEXUS format, which is geared specifically towards commonly used phylogenetic packages such as PAUP*, MrBayes, and BEAST. NEXUS files are composed of different ‘blocks’ that can be added manually by the user or automatically by different software packages. Blocks always use the same syntax:
Example of NEXUS format:
Begin XXXX;
.
.
.
.
.
End;
Note that the semicolons are important! Open up the file rattlesnakes-CMOS-Aligned-AllTaxa.nexus using a text editor on your computer (I recommend BBEdit if available). This file contains an alignment of partial sequences of the nuclear protein coding gene CMOS from several rattlesnakes. You can see that this is a NEXUS file by the block near the top. The ‘Begin data’ block contains general information about the data including the number of taxa (NTAX) and number of characters (NCHAR). Scroll all the way to the bottom and you will see additional blocks of information, one of which is a codon block. As these sequences are part of an exon, nucleotides will belong to one of three codon positions.
The final file type to be familiar with is the PHYLIP format. Like FASTA, this is a relatively simple format that contains two values on the first line—the first indicating the number of taxa and the second representing the length of the sequences. A PHYLIP file containing three taxa and sequences of 15 characters would look like the following:
Example of PHYLIP format:
3 15
Species1 AATGTACCATGGAAT
Species2 AAGGAACCATGGAAC
Species3 AATTGAACCATGGAT
The PHYLIP format was originally developed by Joe Felsenstein for his PHYLIP phylogenetics package, but is today more commonly used for maximum likelihood phylogenetic inference using the programs RAxML-NG (Kozlov et al. 2019) and IQ-TREE (Minh et al. 2020).
Now that we are comfortable with some of the different file types, let’s go back to our GenBank entry for human papillomavirus type 13 coat protein gene (L1), partial cds (Accession M69076.1). Say you are interested in downloading this sequence and using it for phylogenetic analysis. This is easily accomplished by using the ‘Send to’ button in the upper right corner (Fig. 4)
Fig. 4. Sending the data from a GenBank entry to a data file.
Click on Send to > File, and select FASTA Format. This will save a text file containing the sequence. Try opening the file using a text editor (e.g. BBEdit).
This is all fine and dandy, but to perform a phylogenetic analysis we obviously need more than one sequence. Fortunately, GenBank provides an easy way to collect sequences of interest so they do not need to be saved one by one. Go back to the ‘Send to’ button and instead of choosing File choose Clipboard. This will save the entry to the clipboard to be downloaded later. You will see a Clipboard link on the top right (Fig. 5).
Fig. 5. Saving GenBank entries to a clipboard for subsequent use.
Now, you can search other homologous sequences of interest and add each of them to your clipboard. When you click on the clipboard link you will see all the entries that were saved in your search. You can then export all the sequences in a single text file in FASTA format. This is quite handy and something that you will most likely use at some point.
Say you have a sequence of interest, and are looking for other homologous sequences to compare your sequence of interest to. It may be difficult and time consuming to keep typing things in the search box of GenBank. Fortunately, BLAST does the hard work for us. BLAST contains several different alignment algorithms depending upon the type of query sequence and target database to search (Fig. 6).
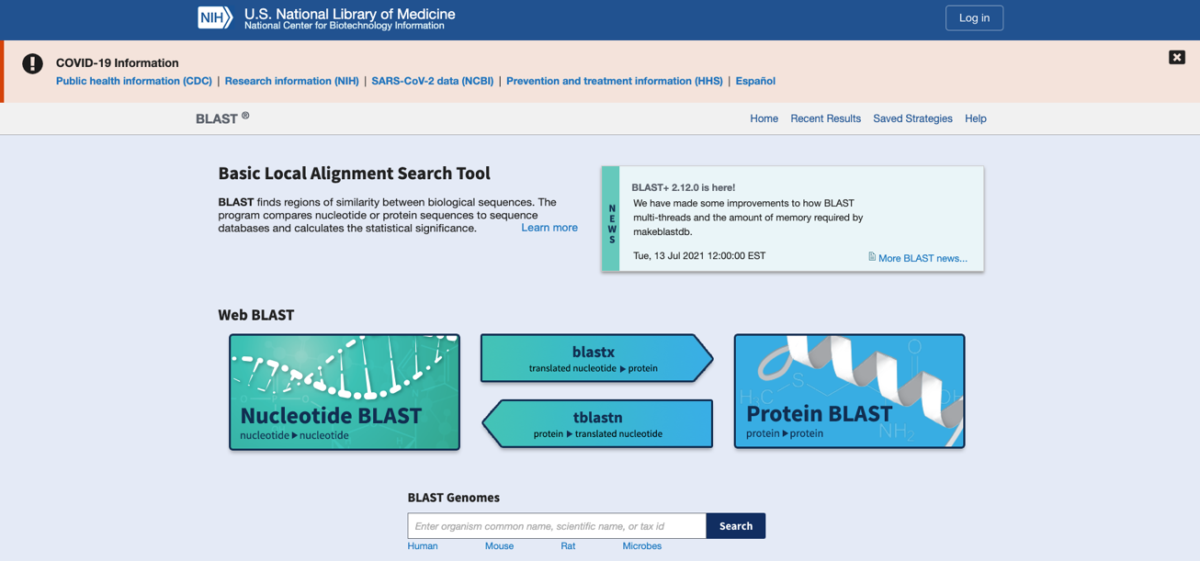
Fig. 6. Alternative versions of BLAST.
Go to the BLAST homepage (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and click on Nucleotide BLAST, and enter the Accession Number MT456249 in the search box. Make sure the Nucleotide collection database is selected and optimize for somewhat similar sequences (blastn). Go ahead and click the blue BLAST button to start the search. When the search finishes you will see an image resembling the following (Fig. 7). Take some time to go through the results to make sure you understand what is being shown.
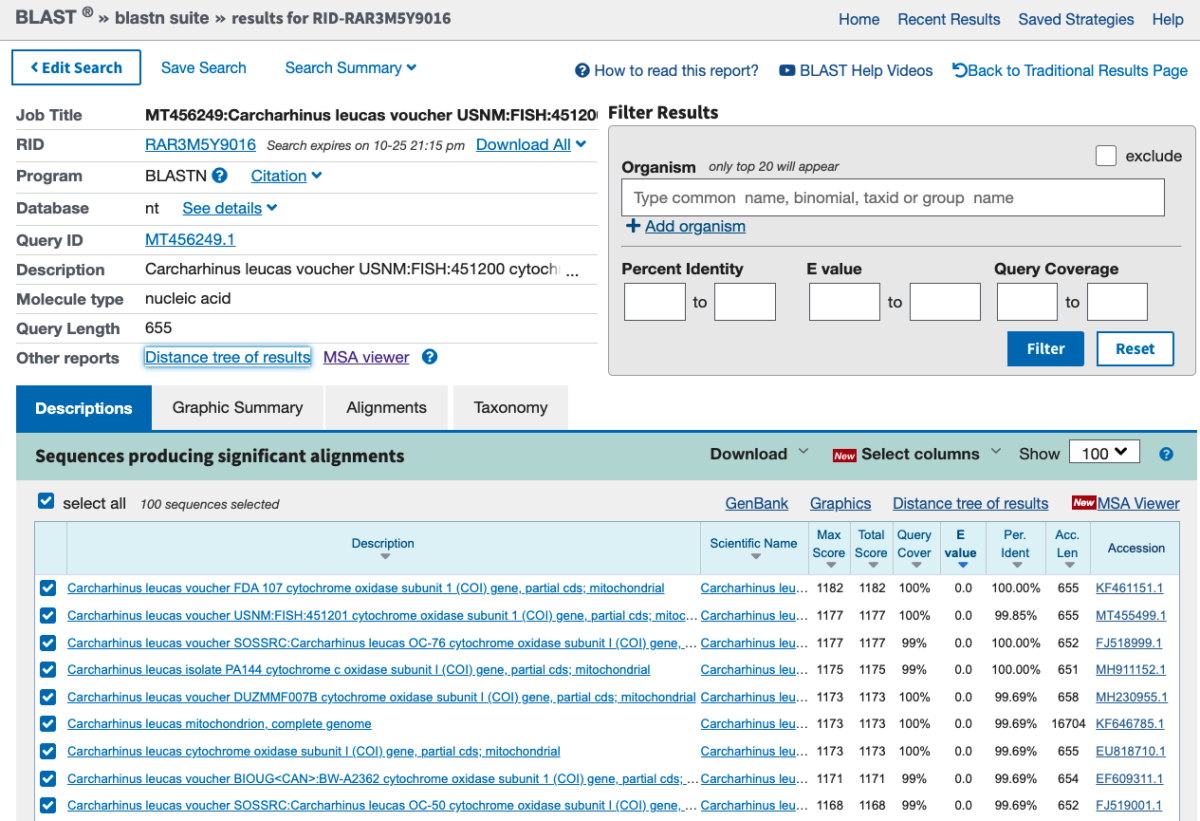
Fig. 7. Results of a BLAST search.
The query sequence is our sequence of interest that we BLASTed (MT456249) and all other rows represent significant hits/alignments. Scroll down and you will see descriptions of the hits, sorted by E() and percent identity. The E-value represents the probability of the match occurring by chance (i.e. not because of true homology). Thus, lower values are more likely to represent similarity in sequences due to shared ancestry. The percent identity column shows how similar the query sequence is to the match. Each entry can be clicked and the data saved as discussed above. If you click on the Alignments tab you will see the actual alignments.
Q: What is the genus and species name for the best match? What is the common name for the organism?
Q: What is the identity of the gene that was queried? Is this a nuclear or mitochondrial gene? Is it a protein coding gene?
Q: What other species are a close match to the query sequence? How did you determine this?
Q: Are all of the best matches from the same genus? If not, what other genera are represented?
Q: What do you think the ‘Query Cover’ term represents?
Multiple Sequence Alignment
Obtaining an accurate multiple sequence alignment (MSA) is imperative if meaningful phylogenetic analyses are to be conducted. The many different alignment algorithms available attests to the interest in the field of methods development. Some of the more accurate MSA methods include MUSCLE (Edgar 2004), MAFFT (Katoh & Standley 2013), and T-COFFEE (Notredame et al. 2000), although Clustal (Larkin et al. 2007) continues to be one of the most popular methods. The programs come as standalone software for download or web versions where you can upload your data for alignment. Downloaded versions can be run from the command line. A MSA may also be created through manual alignment (i.e. by eye). Manual alignment has several pros and cons. In some cases it is easier for a human to determine homology based on what “looks right”. However, when working with highly divergent sequences and/or non protein coding sequences it can be too difficult to align by eye. Aligning by eye is also not feasible when working with genome-scale data sets.
In most cases, we want to actually visualize a MSA and make edits/corrections if necessary. Most MSA algorithms do a decent job providing a good MSA, but sometimes changes are necessary depending on the level of divergence among sequences. Several software packages are available for this purpose. In addition, many of these packages have built-in MSA functions including MUSCLE and/or MAFFT. Two free programs that I like to use are AliView (Larsson 2014) and SeaView (Gouy et al. 2021). These programs also allow you to import/export alignments in different file formats (e.g. FASTA, NEXUS, PHYLIP, etc.).
In this exercise we will work with two data sets, one easy and one difficult. The first data set consists of 18S sequences from several different animal species (animals.fasta). These data were used to help determine the phylogenetic affinities of a strange wormlike organism in the genus Xenoturbella (Fig. 8).
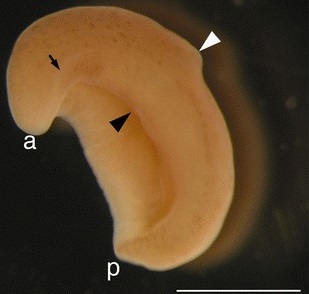
Fig. 8. Image of Xenoturbella bocki. Black triangle indicates mouth. Photo credit: Hiroaki Nakano.
Q: What kind of gene is 18S? Is it a protein coding gene? Is it a nuclear or mitochondrial gene? Why do you suppose that researchers chose to sequence the 18S gene in this project?
The second data set consists of RAG1 sequences from multiple species of rattlesnakes (Fig. 9; Blair & Sánchez-Ramírez 2016).

Fig. 9. Eastern Diamondback rattlesnake (Crotalus adamanteus). Photo credit: Kevin Enge (flickr.com/photos/myfwc/14996160731).
Q: What kind of gene is RAG1? Is it a protein coding gene? Is it a nuclear or mitochondrial gene? Do you think that RAG1 has a fast rate of evolution?
First, open AliView and import animals.fasta.
Q: How many species (sequences) are in the data set? Are the sequences all the same length?
Take some time to try and align some of the sequences manually. Click Edit and turn on edit mode. You can then click on a sequence and move it forward using the spacebar and backward be delete or backspace. Remember that gaps represent insertion/deletion events (indels) and are inserted during a MSA to maintain homology. Try to manually align the two mollusc sequences. Are you able to align these with certainty? Do you know where to insert gaps? It is easy to align the two sequences near the beginning (simply move the Nucula sequence two places the right). However, the alignment should get much more difficult in subsequent positions.
Q: Why do you think these sequences are so divergent? After all, these are all 18S sequences.
AliView is pretty versatile and lets you add/remove taxa, edit sequences, translate nucleotides to amino acids specifying different genetic codes, etc.. Because our 18S sequences will be difficult to align by hand, we will perform automatic alignment. Select all sequences (Selection > Select All) and remove all gaps (Edit > Delete All Gaps in all Sequences). You will notice that the sequences no longer have any gaps (-). Click on Align > Change Default Aligner Program > for realigning all. You should see that the MUSCLE algorithm is already built-in. Click Align > Realign everything and you should have a new MUSCLE alignment. Spend a few minutes scrolling through the alignment to see if MUSCLE did a good job.
Q: How long is your MSA? Are there many indels present? Are there any ambiguous regions of the alignment that you may want to remove? Is the level of variability consistent across the gene, or are certain regions more variable than others?
We can save our MUSCLE alignment by clicking File on the menu. You may choose one of several formats including fasta, phylip and nexus. Save the alignment to your desktop.
Now, let’s try to align the rattlesnake RAG 1 sequences (rattlesnakes-RAG1-Unaligned.fasta). Close the 18S data and open a new AliView window to import the RAG1 data. Similar to the 18S data, take some time to try to manually align the sequences.
Q: Is it easier to manually align the rattlesnake RAG1 data set or the previous 18S data set? Explain your reasoning.
Once you finish manually aligning the rattlesnake data, reopen the unaligned file and perform a MUSCLE automatic alignment.
Q: Is the MUSCLE alignment consistent with your manual alignment? How long is the total alignment? Are there any indels present? Note that gaps and Ns generally represent missing data.
Aliview also allows you to translate nucleotide sequences into amino acid sequences using the correct reading frame and genetic code. Make sure that the Standard Code is selected under View > Select genetic code for translation. Next, starting from your MUSCLE alignment, try translating the nucleotide sequences in different reading frames. Click View > Show as translation, and you should see an image such as the one below (Fig. 10).
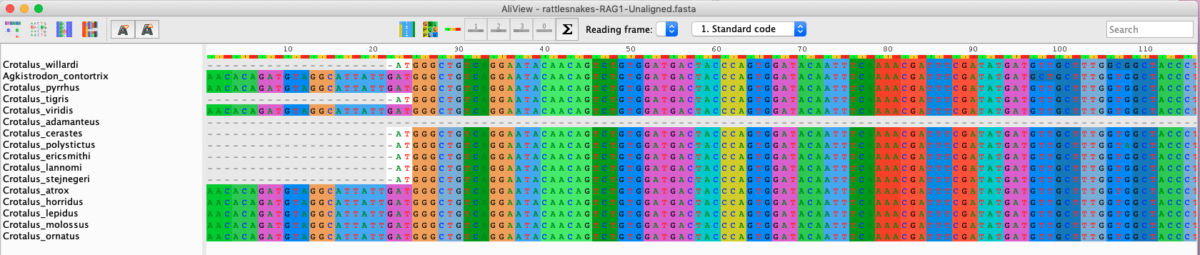
Fig. 10. Rattlesnake RAG1 sequences in translation mode.
Notice that the Standard genetic code is used, which is correct. Our goal is to find the correct reading frame for the translation. For example, should the first nucleotide in the alignment be codon position 1, position 2, or position 3? There are a few ways to do this. In Aliview, click on the sigma icon, which will count the number of stop codons in the alignment. For functional protein coding genes there should be no stop codons present in the middle of the gene.
Q: How many stop codons are present in reading frame 1, reading frame 2, and reading frame 3? Which reading frame do you think is correct?
To make sure we know the correct reading frame, you can BLAST one of the sequences. Click on one of the sequences and then Edit > Copy selection as characters. Paste the sequence into the search box for a nucleotide blast and run the search. Next, click on one of the matches to open up the full NCBI record. Scroll down until you see the translation section that lists the translated amino acid sequence. Does this sequence correspond with your predicted reading frame?
Concatenating several independent MSAs (in class or homework)
AliView is quite useful when working with Sanger sequences and I highly recommend it for your work. Surprisingly, however, AliView is not able to concatenate sequences. In concatenation, one MSA is simply added to the end of another MSA and so forth. Concatenation has been a general practice in systematics and phylogenetics for decades, as more characters/data is generally a good thing! However, there are some important limitations to concatenation, which we will discuss later on in the course. An important thing to remember when concatenating is that the taxon names in each MSA MUST be identical. If they are not, new rows will be added to the MSA. Thus, it is usually necessary to carefully inspect both your individual and concatenated alignments. Several programs allow you to concatenate sequences, my favorite of which is Geneious. However, Geneious requires a license, which can be pricey. Thus, for this exercise we will use the freely available SeaView package (http://doua.prabi.fr/software/seaview).
Go ahead and download SeaView to your system and open it up. For this exercise we will be using two MSAs from rattlesnakes (i.e. rattlesnakes-CMOS-Aligned-AllTaxa.nexus and rattlesnakes-NT3-Aligned-AllTaxa.nexus). Simply click and drag each alignment into separate SeaView windows. The first step is to make sure that the sequences in both files are actually aligned. In other words, always align each gene/locus individually prior to concatenation (this makes alignment much easier). Next, make sure that the taxon names are identical in both files. If not, you can edit them either directly in SeaView or by using a text editor of your choice. The rest is easy. Click on File > Concatenate in one of your MSA windows and make sure that the ‘by name’ circle is checked. Hit OK and watch the magic happen! Your second MSA has now been appended to the first.
How does everything look? Were there any errors? Are the data still aligned? We could do this for multiple genes to create a single long MSA for phylogenetic analysis. When concatenating, it is always a good idea to keep track of which sections of the alignment correspond to which gene/locus and codon position. This will be important when we partition the data for phylogenetic analysis. Also, gaps, Ns, or ?s can be used in a MSA to represent missing data.
Q: Which nucleotide positions in the concatenated alignment correspond to CMOS? Which positions correspond to NT3?
References
Blair C, Sánchez-Ramírez S. 2016. Diversity-dependent cladogenesis throughout western Mexico: Evolutionary biogeography of rattlesnakes (Viperidae: Crotalinae: Crotalus and Sistrurus). Molecular Phylogenetics and Evolution 97, 145-154.
Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32, 1792-1797.
Gouy M, Tannier E, Comte N, Parsons DP. 2021. Seaview Version 5: A Multiplatform Software for Multiple Sequence Alignment, Molecular Phylogenetic Analyses, and Tree Reconciliation. Methods in Molecular Biology doi: 10.1007/978-1-0716-1036-7_15.
Kattoh K, Standley DM. 2013. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution 30, 772-780.
Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. 2019. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453-4455.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947-2948.
Larsson A. 2014. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276-3278.
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R. 2020. IQ-TREE: New models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution 37, 1530-1534.
Notredame C, Higgins DG, Heringa J. 2000. T-coffee: a novel method for fast and accurate multiple sequence alignment. Journal of Molecular Biology 302, 205-217.
Print this page
